Frequently Asked Questions
Table of contents
- Setting the parameters right
- I can’t get an annotated network, despite having correct input files and well-tuned parameters
- Frozen
Sending data to serverpop-up - GTDB and microbetagDB versioning
- How to read a KEGG map with pathway complementarities ?
- I am trying to run
microbetaglocally, but.. - I have a really large 16S-oriented network. What can I do?
- Reconstructing GEMs locally
- Can’t import files on my macOS
Setting the parameters right
Choose input type
It should be clear by now that you always need to load your abundance table. If you wish microbetag to infer a co-occurrence network using FlashWeave then go ahead and set this parameter to abundance_table. In case you already have a network, and you have loaded it to MGG as described in the relative tutorial then you may set this parameter to network so microbetag will annotate the provided network.
What taxonomy scheme to choose ?
For microbetag to return the best annotations it could come up with, it is essential to map as best as possible the sequences described in your abundance table to corresponding GTDB genomes. There are 4 taxonomy schemes supported:
-
GTDB: in case you have used GTDB-tk to taxonomically annotate your bins -
Silva: in case you have used DADA2 along with the 7-level version of Silva they support -
microbetag_prep: in case you have amplicon data and would like to use our implementation for mapping your OTUs/ASVs to GTDB genomes directly by using theidataxaalgorithm of the DECIPHER package and the 16S genes of the GTDB genomes as a reference database. For large datasets (>1000 sequences) see also -
other: in case you want to use your taxonomies and get the closest NCBI Taxonomy names included in the microbetagDB (using thefuzzywuzzyPython library)
When to enable the sensitive and heterogeneous arguments?
Both these parameters are arguments of the FlashWeave software. Therefore, they are only available if you have selected abundance_table as your input type.
The core idea for FlashWeave is to distinguish direct and indirect associations between the taxa of an abundance table. More specifically, for each target variable \(T\) (OTU/ASV or a metavariable), FlashWeave tries to infer its directly associated neighborhood, meaning the set of neighbor variables that renders all remaining variables probabilistically independent of \(T\).
To this end, statistical tests for conditional independence iteratively remove indirect edges. In each iteration a pair of taxa is tested to be directly associated or not and a conditioning set of other taxa is used to test whether the association between the two taxa under study disappears or significantly weakens when conditioning on another taxon.
Apparently, the abundance table is all FlashWeave cares about! The figure below comes from the Supplemental Information of the FlashWeave paper.
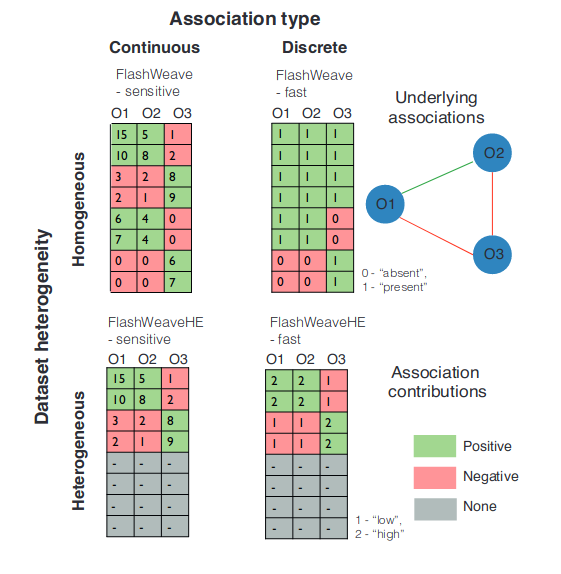
Sensitive modes of FlashWeave use full abundance information (“continuous”), while fast modes (i.e., the sensitive parameter is not selected) work on discretized abundances. As a rule-of-thumb, the sensitive module requires further computing resources and time, thus it may lead the online version of microbetag to time errors. In this case the user can always go for the microbetag_prep steps to infer the network locally. However, in other cases, especially when the number of samples is low, not choosing the sensitive mode can also lead to errors. This is because FlashWeave fails to infer any relationship at all, so there is no network for microbetag to annotate! ![]()
The heterogeneous module (FlashWeaveHE) makes the assumption that zeroes in large, heterogeneous data sets are mostly structural. Thus, it only considers samples in which both OTUs/ASVs have a non-zero abundance as reliable for association prediction. Zero elements are excluded from association computations. However, this restriction only affects the potential association partners being tested: OTUs in the conditioning set keep their absences.
The FlashWeaveHE approach may lose in sensitivity but in large datasets this loss has been found to be rather small, while saving significant computing time. Yet, if applied in small datasets it can also lead to no network inference and therefore microbetag to fail.
What is the Consider children taxa parameter?
This parameter is only valid if the taxonomy database selected is Other. In this case, microbetag tries to use the taxonomies provided to their closest NCBI Taxonomy. There is a chance that your OTU/ASV has been assigned to a species for which a genome is not present in microbetagDB, but genomes of strains of that species are. By enabling this parameter microbetag will consider those genomes and use them for the following annotation steps.
We remind that it is always a good practice in terms of getting as many and as good as possible annotations to run the microbetag_prep step instead of using the Other taxonomy.
I can’t get an annotated network, despite having correct input files and well-tuned parameters
This may be caused because of the time limit of our server for a single run. Thus, there are cases for which even the number of sequence identifiers is less than 1000, combinations of other factors can lead to time-consuming runs, causing an error in the end. For example, you have set taxonomy to other and you have also chosen sensitive as a parameter for the network inference. Such a scenario often leads to time-errors. In this case, you should first run the microbetag-prep step and use its output as your input files. Also, you may have a short number of taxa but a vast amount of samples. In this case, you will also get a time error in case you enable the sensitive parameter of FlashWeave. It is always a good practice to run the preparation step locally, so you also have a better overview of the network you will then ask microbetag to annotate. Remember that microbetag focus is in annotating a network, not building one.
Frozen Sending data to server pop-up
In cases where a time error has occurred, we have observed that from time to time the pop-up box with the progress of your query keeps showing that your data are in process. If that happens, you need to kill the process of the Cytoscape instance. For example, in a Linux system, you would have to check on your htop panel which is the PID for Cytoscape and then run kill <PID>.
GTDB and microbetagDB versioning
GTDB releases a new version once a year, most of the time in April increasing its number of genomes to a great extent from version to version. Even the number of high quality genomes is not increasing that fast, the pairs for microbetagDB to store increases exponentially. We intend to develop a new feature to export new reference genomes and run precalculations over them once a year, following GTDB versioning. Yet, this is still work-in-progress.
How to read a KEGG map with pathway complementarities ?
Complementarities need to always be considered as potential. There is no evidence that just because a complementarity could be occurring based on the genomes of a species pair that is actually happening. To argue about such a case, one would have to get experimental data.
Regarding the pathway complementarities, one need to consider that one pathway does not take into account what is happening to the others. Thus, in cases where both \(pathway_A\) and \(pathway_B\) perform processes that come up with the same end-products, and \(species_A\) has a complete alternative for \(pathway_A\) but not for \(pathway_B\) and \(species_B\) could potentially complete \(pathway_B\), then microbetag would return this potential complementarity, even if it is not necessary for \(species_A\) as it gets what it needs on its own, using \(pathway_A\).
The number of the KOs required for a complementarity to happen (number of KO in the Complement column) is also indicative for its likelihood. If only one KO term needs to be provided by the donor species to the beneficiary, complementarity is more likely to occur compared to a situation where several KOs are required. Also, in case there are also seed complementarities available for a species pair, you can combine information from both types of complementarities. If a seed is close to the pathway mentioned from your pathway complementarities, this adds some extra confidence for the latter to occur.
I am trying to run microbetag locally, but..
When running microbetag locally (see relative tutorial) one may have a wide range of different input files as starting points. You may start with nothing but your bins; i.e., sequencing files, one for each bin mentioned in your abundance table. Otherwise, you may have already annotated them with KEGG ORTHOLOGY terms. You may as well have reconstructed GEMs on your own already. You can adapt your microbetag run by pointing to these files through the config.yml file you have to provide as input. However, we cannot say for sure that no matter the software you used to annotate for example your bins will suit what microbetag expects. If you have a large number of bins, and you would prefer to avoid annotating them again using microbetag this time, you may check the format of the annotations provided in the example case and see if you can edit your format to that. In any case, we strongly suggest you reach out the microbetag’s community on Matrix with any specific questions of yours.
I have a really large 16S-oriented network. What can I do?
There are three things you could do in this case. First, you can try to build a database with the closest genomes you can find for the strains present in you data. If you do so, then you could run microbetag locally using those genomes as they were your bins.
Second, you could build a local instance of microbetagDB locally. This would require a storage of ~700GB. We are now working on an efficient way to go for that.
Third, you can get the annotations per species pairs using the microbetag API. However, in this case, you will not have a .cx file as an end product, i.e. you will not have a single file you can then load on Cytoscape and view through the MGG features.
Reconstructing GEMs locally
There is a great chance when you are trying to reconstruct Genome Scale Models (GEMs) using your own genomes/bins/MAGs and the modelseedpy library, as shown in the relative tutorial, to keep getting messages like:
Recursive run for model_id: /data/my_faa/bin_101
This can lead to excessive time, especially as the number of your genomes increases. This is because modelseedpy requires RAST annotated genomes and thus it needs to establish a connection to the RAST server. Unfortunately, we have observed that this is not always stable.
Can’t import files on my macOS
Make sure you are not using aliases pointing to the files you need to import. Sometimes you may use aliases even without knowing, for example when you drag and drop a file on Finder, you create a shortcut of the file there, but the file is actually located in its original location. Make sure you use the right path and not the shortcut when you are about to import a file on MGG.