ELIXIR Marine Metagenomics Community, initiated amongst researchers focusing on marine microbiomes, has concentrated on promoting standards around microbiome-derived sequence analysis, as well as understanding the gaps in methods and reference databases, and solutions to computational overheads of performing such analyses.
Last month, I had the chance to visit ETH and the Institute of Food, Nutrition and Health.
I was lucky to see the awsome bioreactors they have set over the years there in the Laboratory of Food Biotechnology but most importantly, a group of people that struggle to keep doing what they love.
I was able to see how the data are produced but also spread the world about microbetag, our co-occurrence network annotator.
For sure, two weeks were not enough, so I hope we will meet again even somewhere not as beautiful as the snowy Zurich. A great thanks to Dr. Annelies Geirnaert and Professor for this opportunity and to all the lab members for making my stay so pleasant. Of course, a special thanks to Dr. Andi Erega for all the explanations and patience with me knowing but the basics in the lab but mostly for the burek!
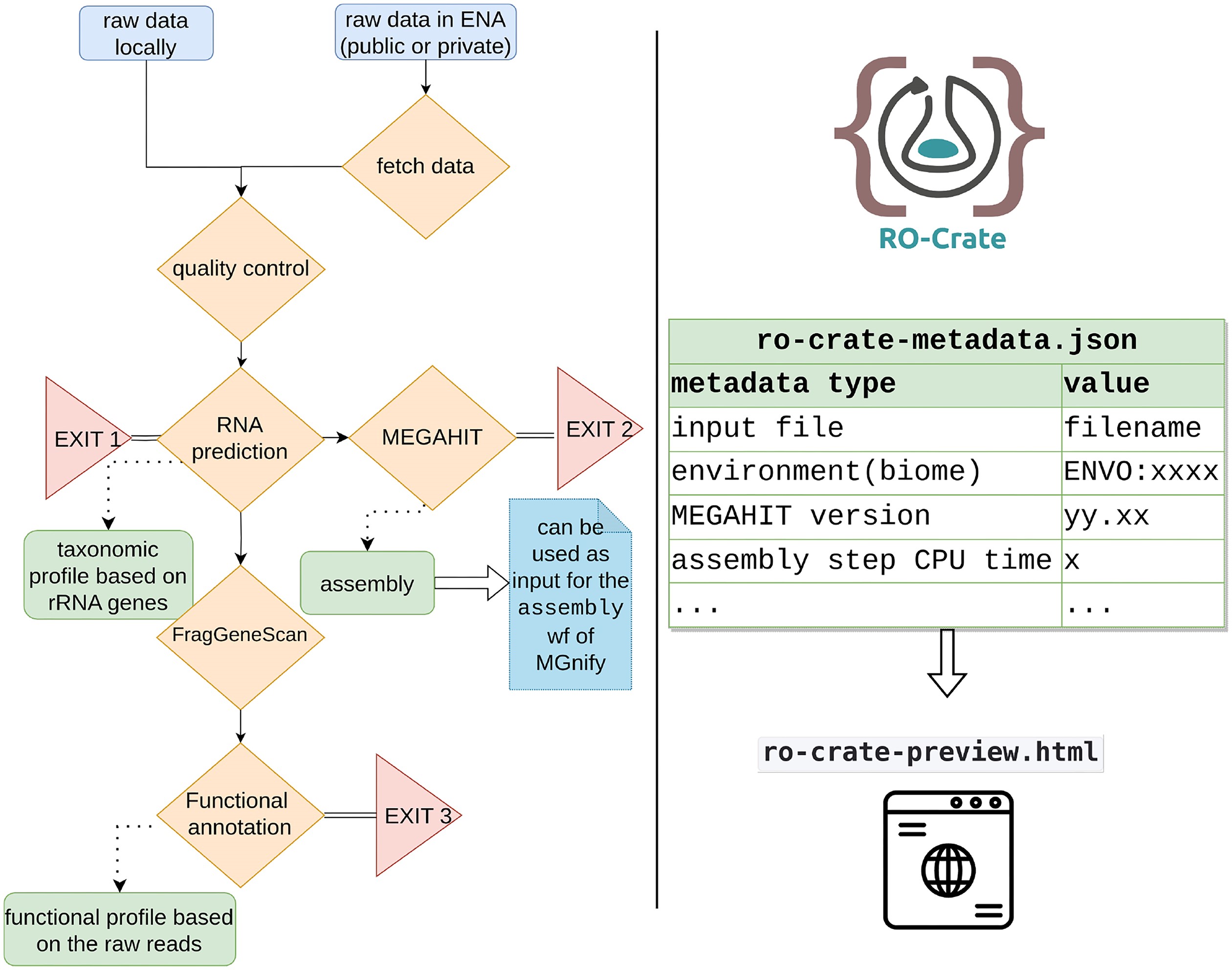
metaGOflow is a Common Workflow Language (CWL) based pipeline for the analysis of shotgun metagenomic data at the sample level. It was initially built to address the needs of the EMO BON community but it can be used for any type of shotgun sequencing data.
metaGOflow is based on the tools and subworkflow implemented in the framework of MGnify.
A multitude of metabarcoding data analysis tools and pipelines have also been developed. Often, several developed workflows are designed to process the same amplicon sequencing data, making it somewhat puzzling to choose one among the plethora of existing pipelines. However, each pipeline has its own specific philosophy, strengths and limitations, which should be considered depending on the aims of any specific study, as well as the bioinformatics expertise of the user. In this review, we outline the input data requirements, supported operating systems and particular attributes of thirtytwo amplicon processing pipelines with the goal of helping users to select a pipeline for their metabarcoding projects.
Based on the established MGnify resource, we developed metaGOflow. metaGOflow supports the fast inference of taxonomic profiles from GO-derived data based on ribosomal RNA genes and their functional annotation using the raw reads. Thanks to the Research Object Crate packaging, relevant metadata about the sample under study, and the details of the bioinformatics analysis it has been subjected to, are inherited to the data product while its modular implementation allows running the workflow partially. The analysis of 2 EMO BON samples and 1 Tara Oceans sample was performed as a use case.
metaGOflow is an efficient and robust workflow that scales to the needs of projects producing big metagenomic data such as EMO BON. It highlights how containerization technologies along with modern workflow languages and metadata package approaches can support the needs of researchers when dealing with ever-increasing volumes of biological data. Despite being initially oriented to address the needs of EMO BON, metaGOflow is a flexible and easy-to-use workflow that can be broadly used for one-sample-at-a-time analysis of shotgun metagenomics data.
Constraint-based approaches have been widely used for the analysis of such models and led to intriguing geometry-oriented challenges. In this setting, sampling uniformly points from polytopes derived from metabolic models (flux sampling) provides a representation of the solution space of the model under various conditions. However, the polytopes that result from such models are of high dimension (in the order of thousands) and usually considerably skinny. Therefore, to sample uniformly at random from such polytopes shouts for a novel algorithmic and computational framework specially tailored for the properties of metabolic models. We present a Multiphase Monte Carlo Sampling (MMCS) algorithm that unifies rounding and sampling in one pass, yielding both upon termination. It exploits an optimized variant of the Billiard Walk that enjoys faster arithmetic complexity per step than the original. Sampling on the most complicated human metabolic network accessible today, Recon3D, corresponding to a polytope of dimension 5 335, took less than 30 hours.