Metabolic models of human gut microbiota: Advances and challenges
in Publications on Fba, Metabolic, Networks
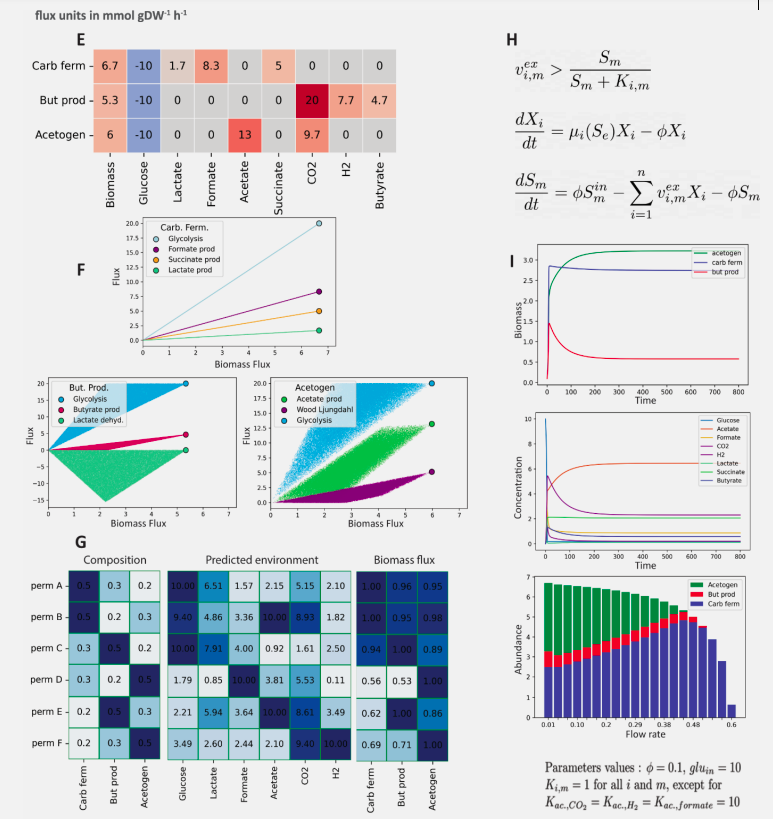
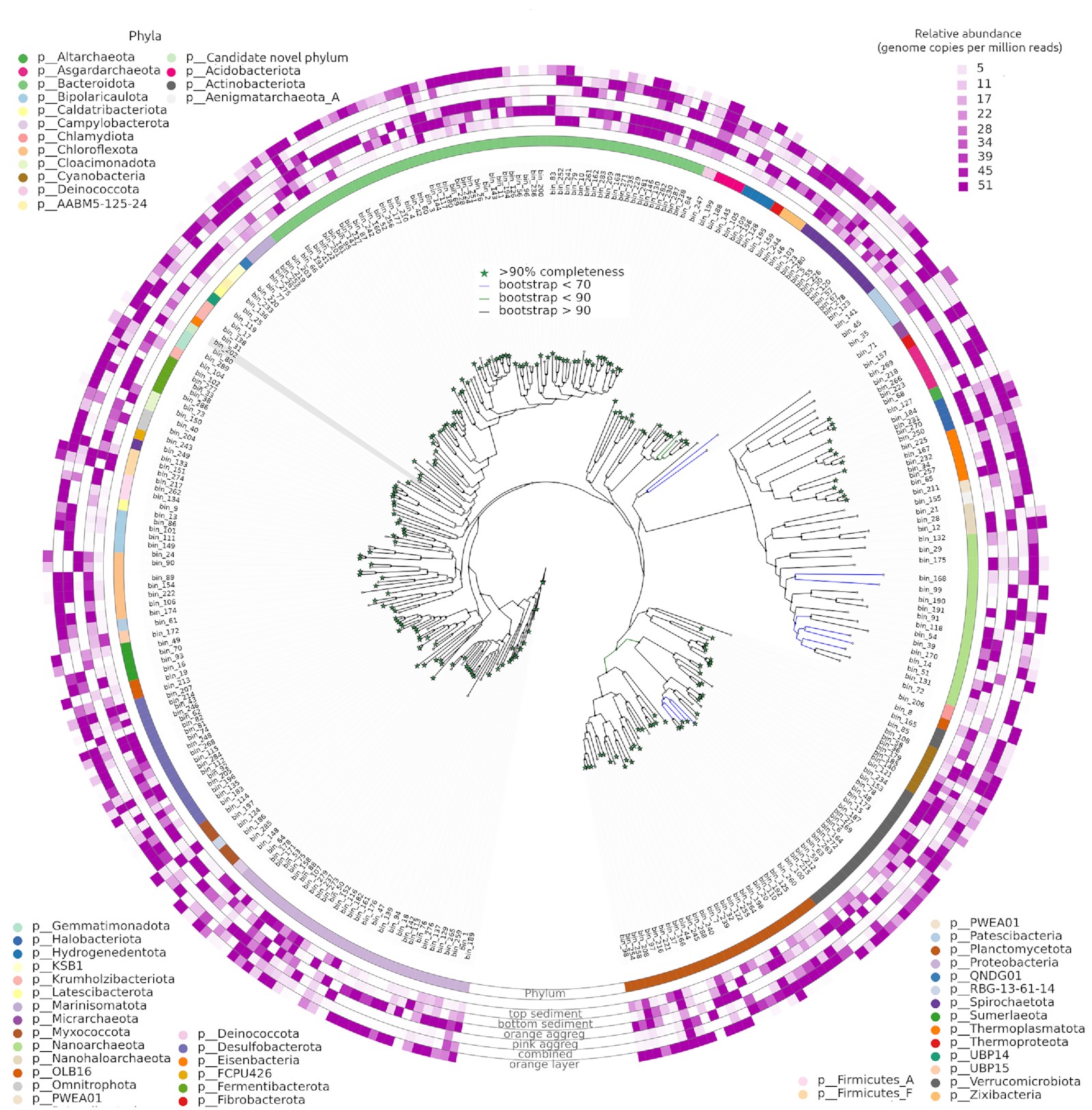
The present study combines 16S rRNA amplicon sequencing and shotgun metagenomics on a hypersaline marsh in Tristomo bay (Karpathos, Greece). Samples were collected in July 2018 and November 2019 from microbial mats, deeper sediment, aggregates observed in the water overlying the sediment, as well as sediment samples with no apparent layering. Metagenomic samples’ coassembly and binning revealed 250 bacterial and 39 archaeal metagenome-assembled genomes, with completeness estimates higher than 70% and contamination less than 5%. All MAGs had KEGG Orthology terms related to osmoadaptation, with the ‘salt in’ strategy ones being prominent. Halobacteria and Bacteroidetes were the most abundant taxa in the mats. Photosynthesis was most likely performed by purple sulphur and nonsulphur bacteria. All samples had the capacity for sulphate reduction, dissimilatory arsenic reduction, and conversion of pyruvate to oxaloacetate.

After 3 years and a half I finally made it! :-)
It’s trivial to say about the good and the bad times, but this is extremely valid.
I will try to keep contact to all those that helped me through this task and to remember what I should do and what I should not.
You may have a look in my Phd-slides:
We have so many things to deal with, war, economic crisis, climate and the list goes on.
Science by itself cannot deal with all of that; it can play a great part thought when focusing on the actual social needs.
Let’s give our best to that direction!

This work focuses on information Extraction (IE) from the marine historical biodiversity data perspective. It orchestrates IE tools and provides the curators with a unified view of the methodology; as a result the documentation of the strengths, limitations and dependencies of several tools was drafted. Additionally, the classification of tools into Graphical User Interface (web and standalone) applications and Command Line Interface ones enables the data curators to select the most suitable tool for their needs, according to their specific features
The high volume of already digitised marine documents that await curation is amassed and a demonstration of the methodology, with a new scalable, extendable and containerised tool, DECO (bioDivErsity data Curation programming wOrkflow) is presented. DECO’s usage will provide a solid basis for future curation initiatives and an augmented degree of reliability towards high value data products that allow for the connection between the past and the present, in marine biodiversity research.

dingo is a Python package that supports a variety of methods to sample from the flux space of metabolic models, based on state-of-the-art random walks and rounding methods. It relies on high dimensional sampling with Markov Chain Monte Carlo (MCMC) methods and fast optimization methods to analyze the possible states of a metabolic network. To perform MCMC sampling, dingo relies on the C++ library volesti, which provides several algorithms for sampling convex polytopes. Among the different ways to sample, dingo also implements the Multiphase Monte Carlo Sampling algorithm (see post for relative publication).
Flux sampling provides insgith of strong statistical evidence. For example, pairwise fluxes correlated with one another in a positive or negative way, can be found.
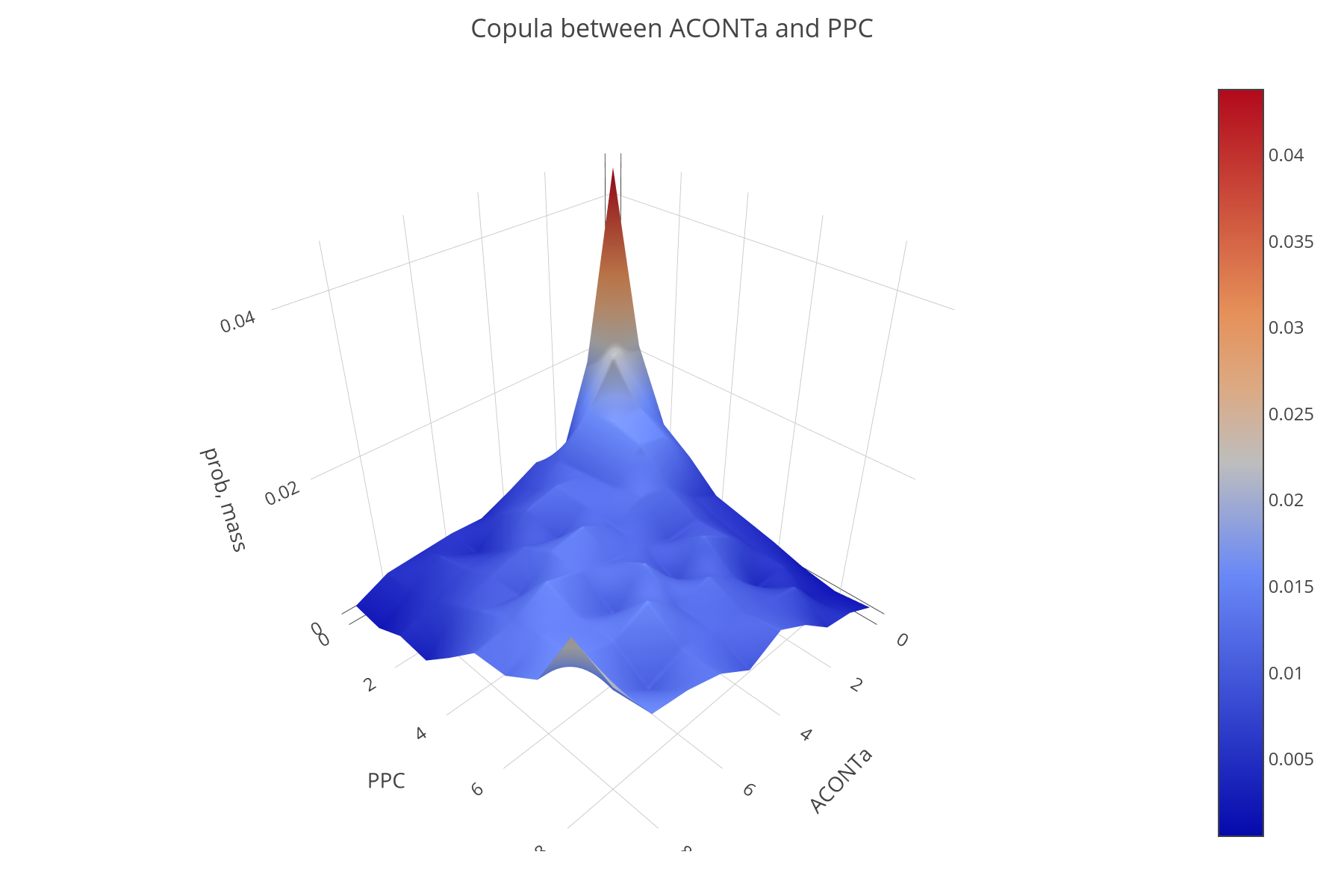
dingo also supports Flux Balance Analysis and Flux Variability Analysis, two standard methods to analyze the flux space of a metabolic network,.
dingo is part of the GeomScale that is over the last year has been an organization of Google Summer of Code.
Really glad to start my EMBO Scientific Exchange Grant in the Lab of Microbial Systems Biology at KU Leuven and the Rega Institute.
Co-occurrence networks have been widely used for inferring microbial associations or/and interactions from metagenomic data. However, spurious associations and tool-dependence confine the network inference. To address this challenge, we are about to develop microbetag, an annotator tool to enhance co-occurrence network analysis for metagenomics data from microbial communities.
To follow our work, you may follow the microbetag GitHub repo.
Many thanks to Prof. Karoline Faust for this opportunity and the incredible feedback!
Let’s see what we can do in 3-months! ![]()
To elucidate ecosystem functioning, it is fundamental to recognize what processes occur in which environments (where) and which microorganisms carry them out (who). Here, we present PREGO, a one-stop-shop knowledge base providing such associations. PREGO combines text mining and data integration techniques to mine such what-where-who associations from data and metadata scattered in the scientific literature and in public omics repositories. Microorganisms, biological processes, and environment types are identified and mapped to ontology terms from established community resources. Analyses of comentions in text and co-occurrences in metagenomics data/metadata are performed to extract associations and a level of confidence is assigned to each of them thanks to a scoring scheme. The PREGO knowledge base contains associations for 364,508 microbial taxa, 1090 environmental types, 15,091 biological processes, and 7971 molecular functions with a total of almost 58 million associations. These associations are available through a web portal, an Application Programming Interface (API), and bulk download. By exploring environments and/or processes associated with each other or with microbes, PREGO aims to assist researchers in design and interpretation of experiments and their results. To demonstrate PREGO’s capabilities, a thorough presentation of its web interface is given along with a meta-analysis of experimental results from a lagoon-sediment study of sulfur-cycle related microbes.
Read more for the PREGO software
With Dr. Christina Pavloudi and Dr. Ioulia Santi we prepared a class for amplicon analysis for starterts.
So, here is a place to start with if you are interested in that:
Our software tool on investigating COI amplicon data just got published on MBMG journal. We hope that darn will benefit researchers as a quality control tool for their sequenced samples in terms of distinguishing eukaryotic from non-eukaryotic OTUs/ASVs, but also in terms of understanding the known unknowns.
🆕Dark mAtteR iNvestigator (DARN) to serve as a quality control tool for sequenced samples in terms of distinguishing #eukaryotic from non-eukaryotic OTUs/ASVs➕understanding the known unknowns.
🔗Software Description: https://t.co/MsYpkyCAp9 #Metabarcoding #eDNA #Sequencing pic.twitter.com/t348UiYsUH— MBMG Journal (@MBMGjournal) November 3, 2021
You may find it on this GitHub repository.
![]()
