PEMA investigating metabarcoding
16/18S analysis
PEMA supports multiple options for running 16S or/and 18S rRNA data.
Clustering OTUs / ASVs inference
You may choose between OTUs clustering or ASVs inference ;PEMA supports both.
On the parameters.tsv file, you will find the clusteringAlgoFor16S_18SrRNA parameter. By setting this as algo_vsearch PEMA will use the vsearch algorithm to get OTUs.
Otherwise, by setting this as algo_swarm PEMA implements the swarm algorithm to infer ASVs.
Silva version 128 and 132
Silva includes two different Silva versions you may choose.
Silva versions besides the increased number of available sequences, they may include different taxonomic frameworks.
You may furhter check on that on the corresponding literature, for example here or visit the Silva database website.
Alignment or phylogeny based taxonomy assignment
For the taxonomy assignment step, PEMA supports two ways when it comes to 16S/18S rRNA data.
By setting taxonomyAssignmentMethod as alignment, PEMA will use the CREST algorithm and the Silva version you selected and will return you an alignment based taxonomy assignment.
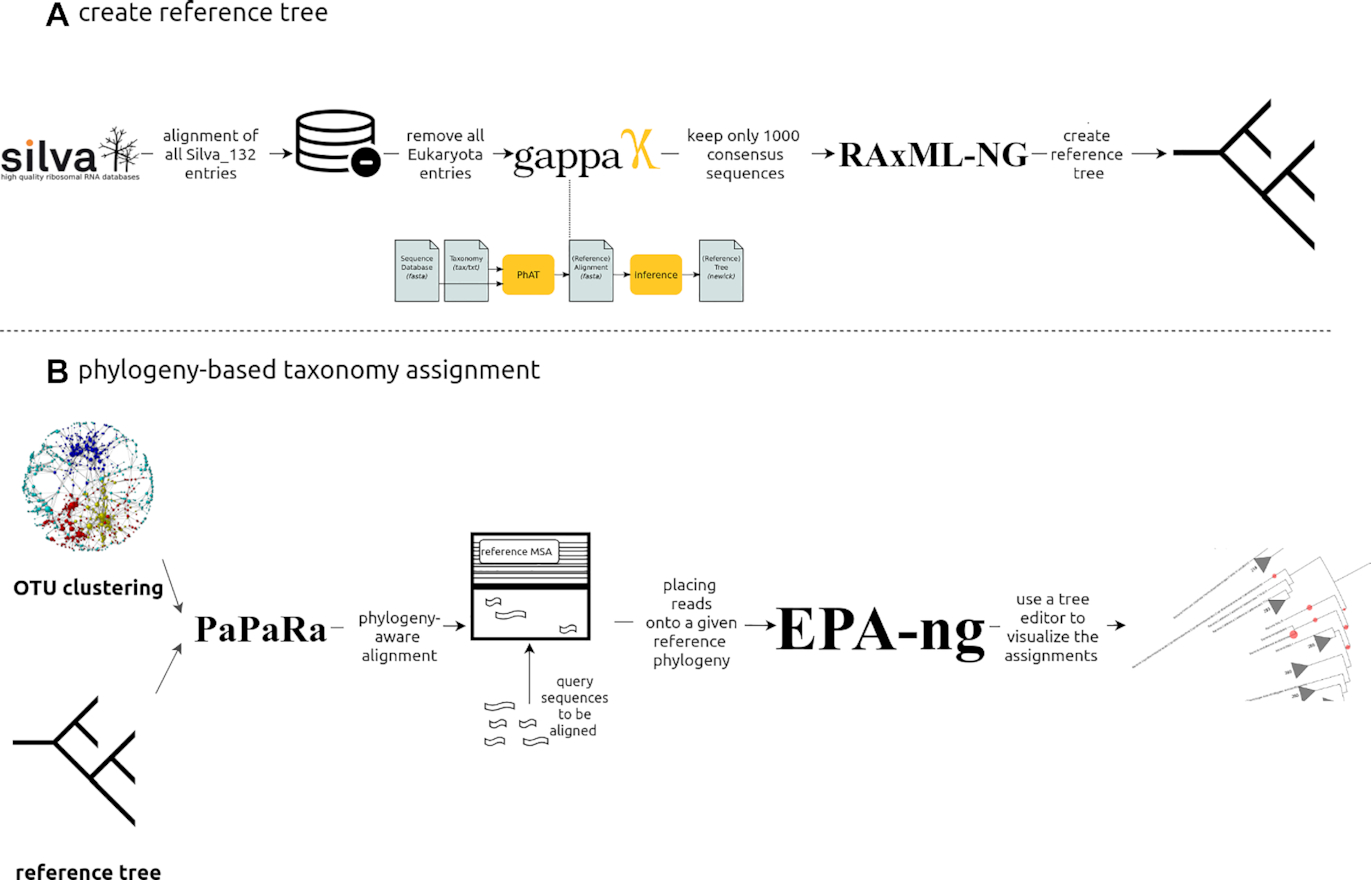
On the contrary, by setting this parameter as phylogeny PEMA will use a reference tree of 1.000 consensus taxa we have built by using Silva 128, the RAxML-ng and the EPA algorithms to return you a phylogeny based taxonomy assignment which you will be able to view via tools such as iTOL.
Here is the workflow we used to build PEMA’s reference tree.
phyloseq analysis
You may ask for PEMA to move on after the taxonomy step and run a phyloseq analysis. Phyloseq is a R package for further exploring microbiome profiles.
To do so, you need to set the phyloseq parameter in the parameters.tsv file as Yes and add the phyloseq_in_PEMA.R script in the analysis_directory with all the rest of your input~
You may get a version of this script here. As mentioned on the script, the first sections need to be exactly like this.
However, you may add or remove phyloseq modules according to your needs.
You always need to remember the metadata.csv file, you may find an example here.
You need to add your own metadata there keeping the format of the file as it is!
You always have to keep the names of these scripts exactly as they are, otherwise PEMA will return you an error!
Plots and all the phyloseq-related output will be saved in a seperate subdirectory called phyloseq_output.